De Novo Pipeline
De novo pipeline building requires bioinformatics expertise. Please contact the technical support team for assistance.
The creation of a brand new pipeline is more challenging than copying an existing pipeline. Fill in the following details on the pipeline creation window (Fig. 1) to create a new pipeline. Mandatory fields are indicated with asterisks (*).
- Name* - Provide a unique name
- Execution Flow* - Enter the tool names in the order of execution (e.g. Trimmomatic -> Salmon -> Sleuth)
- Hub* - Indicate the pipeline group
- Category* - Select the functional category
- Description* - Provide a brief description of the pipeline’s general purpose
- Details - Provide detailed information about tools, inputs, outputs, arguments, and other pertinent information
- Steps* - Step field helps to add tools and commands to a pipeline.
At least one step is required for a functional pipeline.
Click the icon to add a new step (or tool) to the pipeline(Fig. 1). There are eight fields in each step:
- Number - The number determines the step number in the pipeline. It is automatically filled when a new step is added.
HINT: Only positive integers are allowed
- Name - Provide a name to the step (e.g. Bowtie2 alignment). The outputs will override if the name field is not unique because the name is used as a directory to store the output files of the step.
HINT: The name should be unique.
- Tool - Select a tool from the drop-down menu
- Command - Select a command from the drop-down menu for the selected tool
- Predecessor - The number determines the dependency step of a step. A step executes only after the dependency step.
HINT: Only positive integers are allowed
- Merge - This is a critical variable that indicates if all the inputs need to be combined into a single output file. This is useful in some analyses where all files need to be analyzed together (Examples: Differential gene expression and Joint genotyping). Default: No.
HINT: The first step can’t be a merge step
- Input Source - Determines the source of the input files for a step. Typically, input file sources are either Data Store (Sequence Data, References, Annotations, and Metadata) or output files from predecessor steps.
HINT: Input sources from multiple steps are allowed. (Example: BAM and BAI files created in different steps required for Variant calling).
HINT: Currently, Data Store is allowed for the first step only
- Actions - Each step allows the following actions
- Access the Command builder dialog box
- Delete a step
- Copy a step
- Command Builder
-
- Command building is one of the complex processes on the platform and the Command Builder dialog box helps to navigate the process easily.
Commands are preconfigured by the platform admin. Users can only edit the commands.
-
- During the pipeline development stage users define the commands and the final executable command will be dynamically created during the pipeline execution stage.
This is a generic command building process. You are NOT making the actual file selections required for the analysis. The platform does it automatically based on your definitions.
-
- Command Builder has two modes: View mode and Edit mode. The former allows viewing and the latter allows command modification, as described below. Access the Command Builder dialog box (Edit mode) (Fig. 1) by clicking under the Actions column.
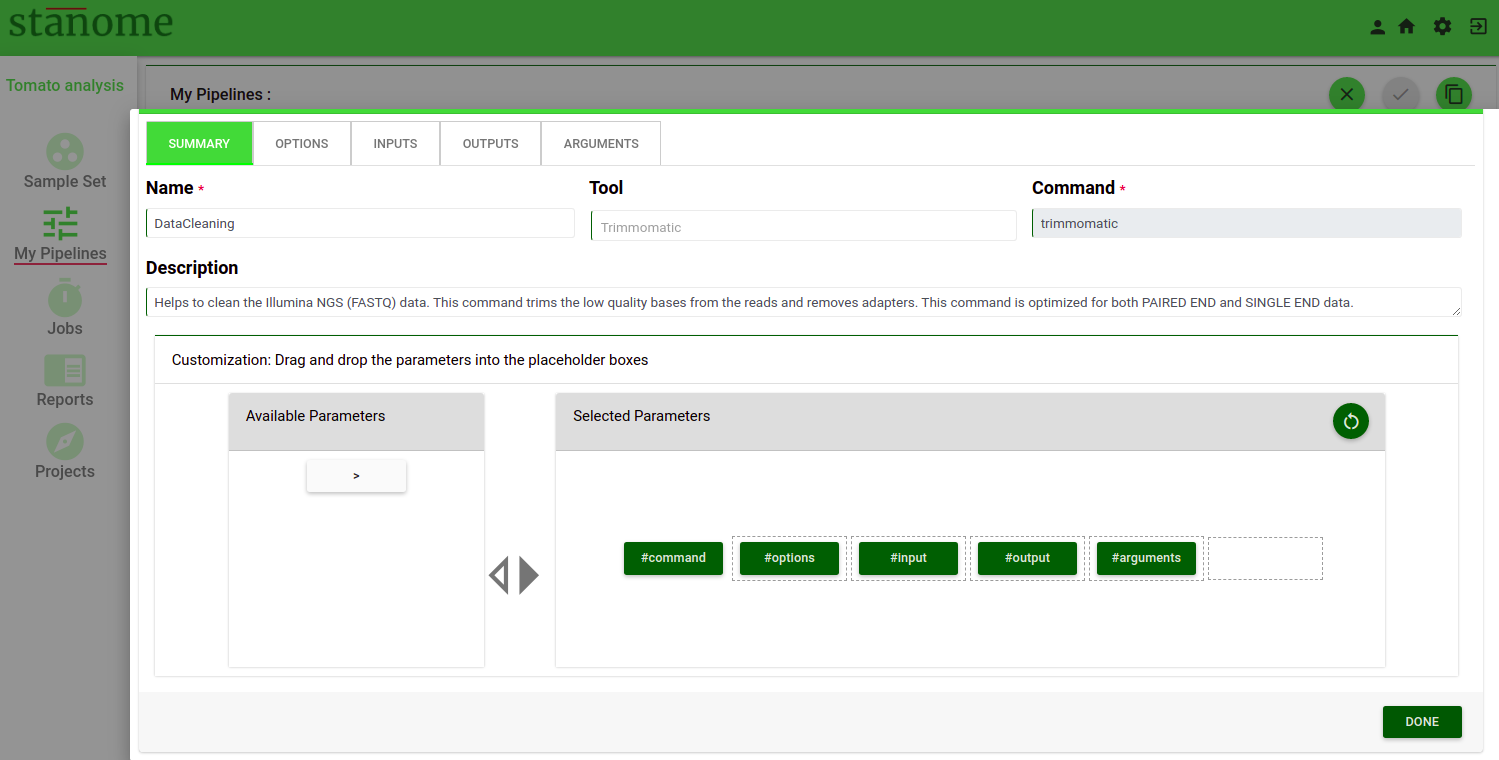
The first tab of the Command Builder describes the generic details(summary) about a command.
-
-
-
- Name - The step name as given by the user while creating the step.
- Tool - The selected tool (cannot be modified)
- Command - The actual command to be executed (cannot be modified)
- Description - Brief description of the command (auto-filled but can be modified by the user)
- Build Your Command - This box helps to build the actual command. The left box shows the available parameters and the right box (Fig. 2) shows the selected parameters. The order of the parameters is extremely important and should be maintained for the command to execute. All commands come with a default parameter sequence (Stanome defined). The parameters are prefixed with a #(hash). The default pattern can be modified by dragging and dropping the parameter buttons (green color) between the left and right boxes. Based on the selection the parameter tabs are enabled on the top.
-
-
Default pattern: #command #options #arguments #input #output
The pattern should ALWAYS start with #command and can’t be edited.
Allowed character: Parameter words, #, space, and >
“>” is allowed preceding the #output ONLY
The second tab of the Command Builder (Fig. 2) describes the Options parameter. Details of the Options tab are described below:
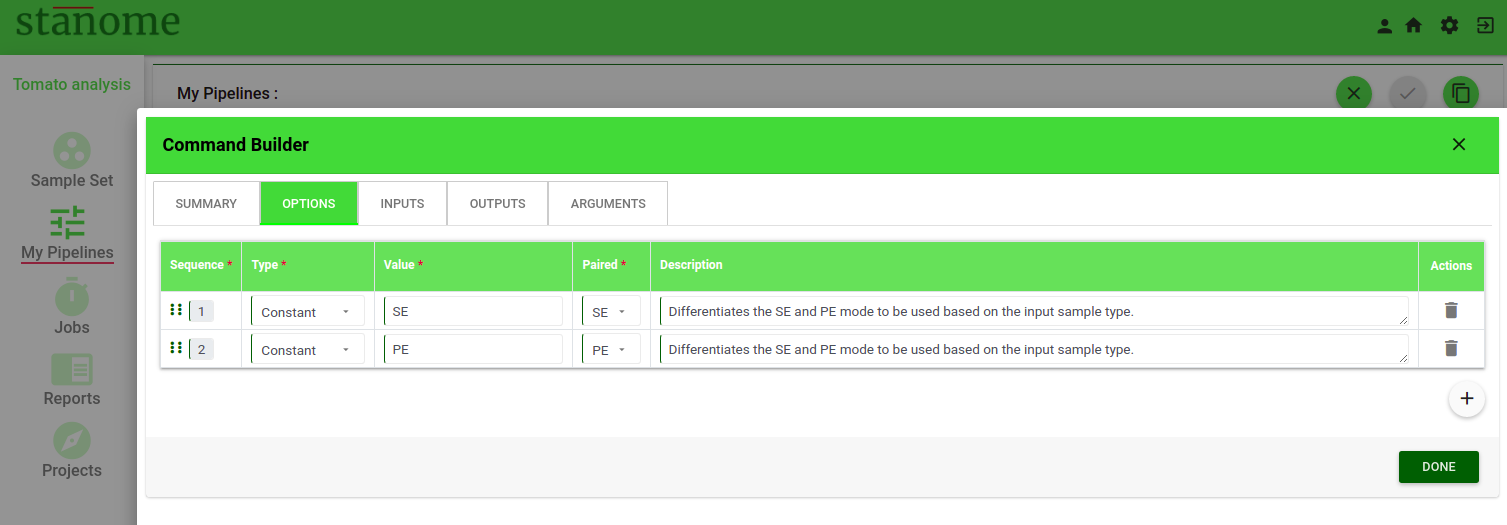
- Options
Single-word parameters should be defined as options (Examples: --ignore, --1, PE, SE). All the options are listed in a table format. New row(s) can be added using the ‘+’ sign at the bottom of the table. Six fields are available under each option.
-
-
- Sequence: This number determines the order of the option in the command
- Type: Six choices are available in the drop-down. Select the type of option. (Example: Annotation, Constant, Metadata, Reference, Threshold, and Variable)
- Value: Based on the field Type, the corresponding values in the drop-down change. Select an appropriate value. See the table below for available field types and their values.
-
CAUTION - Verify usage of each option before using
|
Field Type |
Value |
|
Annotation |
|
|
Constant |
|
|
Metadata |
|
|
Reference |
Define references to select
|
|
Threshold |
Define threshold values to use
|
|
Variable |
Native variables of the platform
|
Table. Available Field Types and their corresponding Values.
-
- Paired: Indicates if an option can be used for paired-end, single-end files, or both (Default: All)
- Description: A brief description of the option functionality or usage guidelines.
- Actions: Allows the deletion of an option.
CAUTION - Please refer to the Arguments section for defining the parameters with key-value pairing
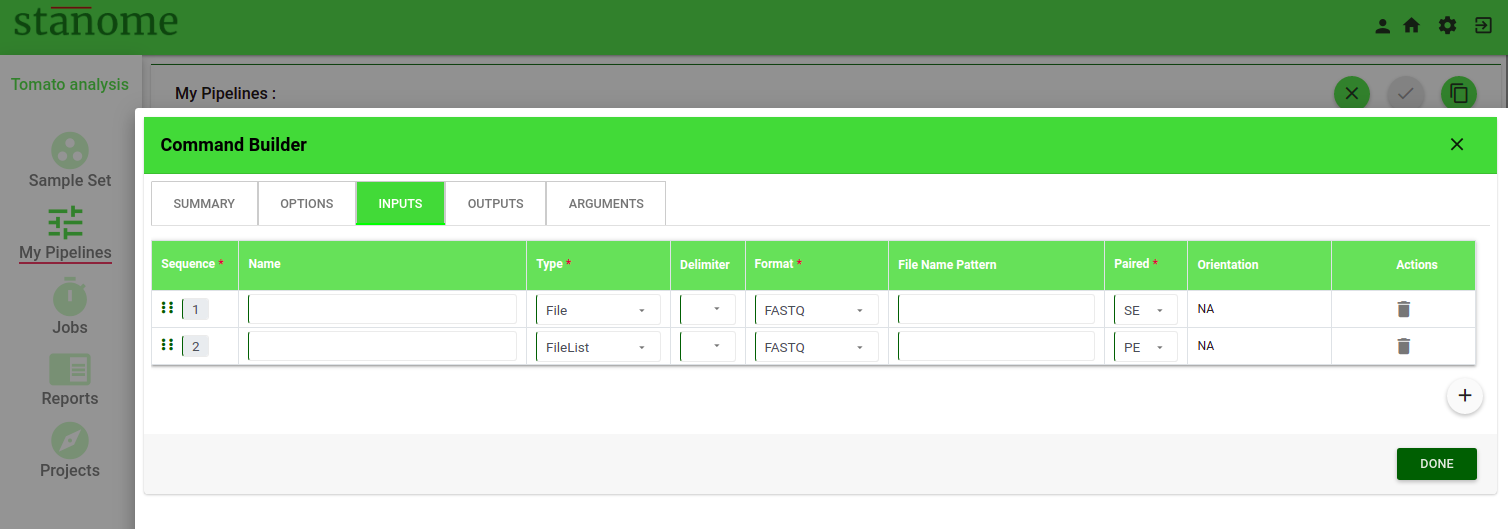
- Inputs and Outputs
Input and output files are defined under INPUTS and OUTPUTS tabs, respectively. Eight fields are available under each of these parameters (Fig. 3).
-
- Sequence: This number determines the order of the inputs or outputs in the command
- Name: The name of the value (Examples: --input, -output)
- Type: Input/output data can be provided to a command in three formats - File, FileList, and Directory. Select the format from the drop-down list.
-
- File - Input is a single file (Example: Prefix.fastq)
- FileList - Input is Paired-end files (Example: Prefix_R1.fastq, Prefix_R2.fastq)
- Directory - Input is a directory path
-
- Delimiter: Character separating the input/output file(s) from its name in the command (Example: --input : Prefix.fastq).
CAUTION - Allowed delimiters are =, -, :, and ;
-
- Format: Depending on the data select file extension from the drop-down. This value is used to make the right file selections during the pipeline execution. Required for File and FileList types only and not required for Directory type.
CAUTION - The file extensions should be precise; even the FASTQ and FQ are treated distinctly.
-
- File Name Pattern: This field is applicable for the Inputs parameter only. Regular expressions can be used to select specific input files. This value is used in combination with the Format field. Few examples are provided below for an easy understanding of regular expression usage.
-
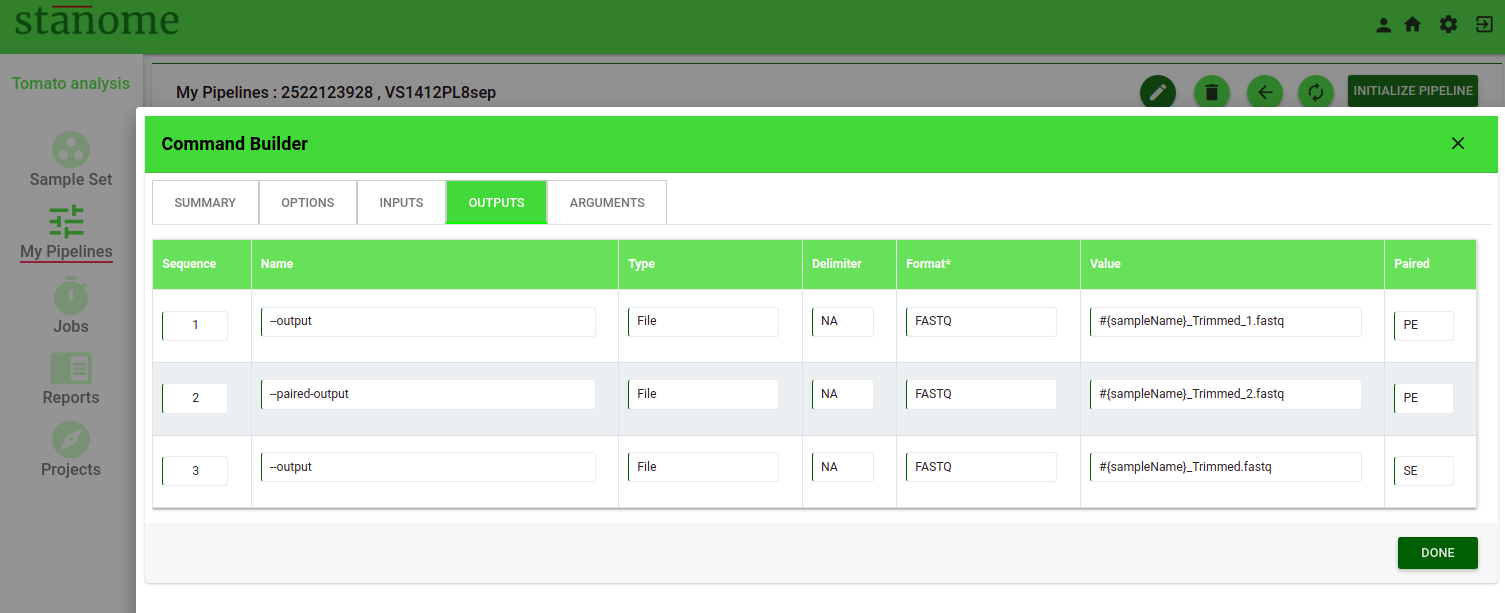
- Value: This field applies to the Outputs parameter only. Output value contains three parts
-
- Prefix: Stanome sample variable (${sampleName})
- Suffix: Step name
- File extension
-
- Value: This field applies to the Outputs parameter only. Output value contains three parts
(Example: ${sampleName}_trim.fastq for trimmomatic step). This helps track the files across the entire pipeline execution.
-
- Paired: Indicates if the files (Inputs/Outputs) parameter is applicable to paired-end, single-end files, or both (Default: All).
- Actions: Allows the deletion of an input or output.
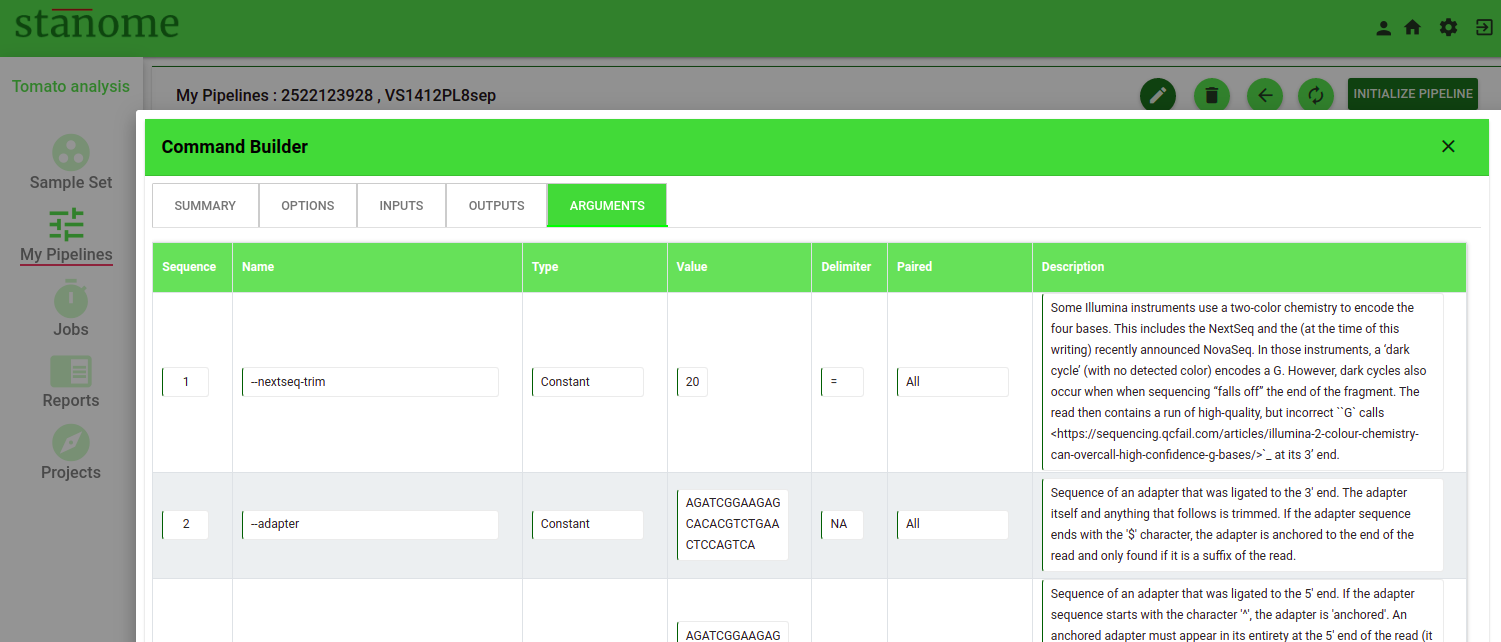
- Arguments
Parameters defined as a key-value pair should be defined as arguments (Fig. 4). Arguments can be used for any parameters supported by the tools and other required files (reference files, gtf or annotation files, target or hotspot files). They are defined by the following eight features:
CAUTION - Please refer to the Options section for defining the singleton parameters
-
-
-
- Sequence: This number determines the order of the argument in the command.
- Name: Name of the argument used by the command to identify it
-
-
Arguments are grouped into categories to support diverse tools and commands. In arguments, two fields (Type and Value) work together to define an argument.
-
-
-
- Type: Seven choices are available in the drop-down. Select the type of argument. (Example: variable, constant, and annotation_DNAseq)
- Value: Based on the Type selected, the values in the drop-down change. Select the appropriate value. Refer to the table given in options for the available types and their values.
- Delimiter: Character separating the keys and values in the command (Example: --count: 10). Not all arguments require delimiters between the Name and the Value fields.
-
-
CAUTION - Allowed delimiters are =, -, :, %, and ,
-
-
-
- Paired: Indicates if the arguments parameter applies to paired-end, single-end files, or both (Default: All).
- Description: A brief description of the argument describing the function and utility of the argument.
- Actions: Allows the deletion of an argument.
-
-
Click on the bottom right corner to save the changes to the command. This is the completion of the first step in the pipeline. Continue adding all the steps until the pipeline is complete. Steps can be dragged and dropped at any position with the icon. Step number, predecessor, and input source get automatically readjusted for all the steps. Click to save the pipeline.